
EFFICACY & SAFETY OF PHARMACOKINETICALLY-DRIVEN DOSING OF MYCOPHENOLATE MOFETIL FOR THE TREATMENT OF PEDIATRIC PROLIFERATIVE LUPUS NEPHRITIS - A DOUBLE-BLIND PLACEBO CONTROLLED CLINICAL TRIAL (PLUMM)
Protocol Synopsis
A. BACKGROUND AND RATIONALE
The safety and effectiveness of mycophenolate mofetil (MMF) for the treatment of lupus nephritis (LN) has been tested in adult and some pediatric subjects. MMF, a prodrug, is rapidly converted to the active moiety mycophenolic acid (MPA) and has been found to be of significant benefit to patients after renal transplant and with LN. Exposure to MPA is associated with the immunological and anti-inflammatory response of the patient. However, the pharmacokinetics of MPA are characterized by large inter- individual and intra‐individual variability. Factors contributing to this variability include differences in albumin concentrations, corticosteroid use, impaired renal function, altered hepatic function, and genetic polymorphisms in drug metabolizing enzymes and drug transporters.
The Sponsor is proposing to conduct a 1-year 2-part double-blinded placebo controlled 2-arm clinical trial. Treatment arms are (1) MMF dosed as per body-surface area (MMFBSA; 600mg/m2 body surface area per dose about every 12 hours) and (2) pharmacokinetically-guided precision-dosing of MMF (MMFPK; MMF dosed twice daily to achieve an area under the concentration-time curve (AUC0-12h) of MPA
>60-70 mg*h/L. The study goal is to determine the safety and efficacy of MMFPK compared to MMFBSA for the treatment of proliferative LN in subjects 8 to <21 years.
Upon enrollment in this study, subjects will be randomized 1:1 to receive blinded treatment with MMFPK or MMFBSA for up to 53 weeks. The primary endpoint, partial clinical remission (PRR) of LN, is measured at the end of Part 1 at week 26. Subjects in the MMFBSA arm who have only achieved PRR at the end of Part 1 will newly receive MMFPK upon entering Part 2 of the study (week 26 – 53). Subjects with complete renal responses (CRR) at the end of Part 1 will continue the same dosing regimen of MMF (MMFBSA or MMFPK) in Part 2 as was given in Part 1 of the study. Subjects in the MMFPK arm with PRR at the end of Part 1 will enter Part 2 and continue in the MMFPK arm.
Subjects who are LN non-responders by the end of Part 1 at week 26 will be considered treatment failures and discontinued from the study intervention. All subjects who are discontinued from the study intervention for reasons of efficacy or safety will receive LN treatment and monitoring as per the treating physician’s decision. However, these subjects will be asked to participate in study visits at weeks 26 and 53/ End of Study.
B. STUDY OBJECTIVES AND ENDPOINTS
Primary:
To compare the efficacy of MMFPK therapy to the efficacy of MMFBSA therapy as measured by the percentage of subjects achieving at least partial remission of LN (PRR) as per the adapted ACR/EULAR Criteria at Week 26 (end of Part 1) of the study.
Major secondary:
To compare the efficacy of MMFPK therapy to the efficacy of MMFBSA therapy as measured by the percentage of subjects achieving complete remission of LN (CRR) as per the adapted ACR/EULAR Criteria at Week 26 (end of Part 1) of the study.
Other secondary:
To compare the efficacy of MMFPK therapy to the efficacy of MMFBSA therapy during all parts of the study (baseline to week 53) as measured by the percentage of subjects achieving CRR;
Exploratory:
To compare the efficacy of MMFPK therapy to the efficacy of MMFBSA therapy during all parts of the study (baseline to week 53) as measured by
a. Change from baseline in the levels of biomarkers of LN activity as summarized by the Renal Activity Index for Lupus (RAIL) score;
b. Cumulative exposure to oral corticosteroids;
c. Cumulative exposure to intravenous corticosteroids;
d. Mean time-adjusted average extrarenal disease activity;
e. Time to CRR;
f. Time to PRR;
g. Frequency of subjects meeting discontinuation criteria;
h. Change from baseline in the health-related quality of life (HRQoL).
To compare the tolerability and acceptability of MMFPK therapy to the efficacy of MMFBSA therapy during all parts of the study as measured by
a. Percentage of subjects who are adherent to MMF as measured by blister-pack count;
b. Percentage of subjects with MPA levels of < 0.10 mg/L as measured during study visits;
To explore the importance of patient age, race and/or gender on the efficacy, safety and tolerability of MMFBSA and MMFPK during the study.
Safety:
To compare the safety of MMFPK therapy to the safety of MMFBSA therapy during all parts of the study as measured by
a. Frequency of adverse events (AE) grade 3 or higher;
b. Frequency of serious AEs (SAEs);
c. Frequency of serious infections.
Endpoints Primary endpoint:
Achievement of at least PRR at the end of Part 1 (week 26) of the study.
Major secondary endpoint:
Achievement of CRR at the end of Part 1 (week 26) of the study.
Other secondary endpoints:
Achievement of CRR at the end of Part 2 (baseline to week 53) of the study;
Exploratory endpoints:
Change in Renal Activity Index for Lupus (RAIL), pediatric algorithm during the study (baseline to week 53);
Cumulative dose of oral corticosteroids (CS) during the study (baseline to week 53); Cumulative exposure to intravenous CS during the study;
Mean time-adjusted score of the Systemic Lupus Erythematous (SLEDAI) considering extra-renal domain items during the study (baseline to week 53);
Time to CRR during the study (baseline to week 53); Time to PRR during the study (baseline to week 53);
Frequency of patients fulfilling discontinuation criteria during the study (baseline to week 53);
Change in HRQoL as measured by the PedsQL Generic Core Scale during the study (baseline to week 53);
Percentage of patients with intake of > 80% of prescribed MMF doses as per blister-pack count during the study (baseline to week 53);
Presence of MPA- levels <0.1 mg/L from random testing or in clinic testing during the study (baseline to week 53);
Safety Endpoints:
Frequency of Grade 3 and higher AEs as per the Common Terminology Criteria for Adverse Events (CTCAE) Version 5.0 during the study (baseline to week 53);
Frequency of SAEs during the study (baseline to week 53); Frequency of serious infections (baseline to week 53).
C. ELIGIBILITY CRITERIA
Inclusion Criteria
Subjects must meet all of the following inclusion criteria at screening and baseline to be eligible for randomization into the study:
1) Male or female aged 8 to < 21 years;
2) Must meet Classification Criteria for SLE as per the criteria of the American College of Rheumatology (ACR)/ European League Against Rheumatism 1-3 (Appendix 0);
3) Diagnosed with proliferative LN as per the International Society of Nephrology/Renal Pathology Society4 based on kidney biopsy done within 90 days prior to enrollment into the study;Subjects may have been previously diagnosed with LN. For study inclusion, the kidney biopsy must be interpreted as one of the following classes: Class 3, Class 3/5, Class 4, or Class 4/5.
4) Treatment of LN with twice daily MMF as per the decision of the treating physician.
The subject will have taken MMF as prescribed by their treating physician for a minimum of 4 days (or 8 doses).
5) Subject tolerates MMF as per the treating physician’s opinion;
6) Able to swallow MMF tablets and capsules;
7) If subject is treated with belimumab, must be IV or SQ;
8) Subjects who are willing and able to comply with scheduled visits, treatment plan, laboratory tests, and other study procedures;
9) Evidence of a personally signed and dated Informed Consent document and Assent document (as appropriate) indicating that the subject and a legally acceptable representative/ parent(s)/legal guardian has been informed of all pertinent aspects of the study.
10) Parent or legal guardian must have a smart phone available and able to support the PLUMM smart phone application.
11) Must be able to complete study questionnaires in English or Spanish.
Exclusion Criteria
Subjects with any of the following characteristics/conditions at screening and baseline will not be included in the study:
1) Perceived or stated inability to adhere to the study protocol;
2) Hypersensitivity to MMF or any component of the drug product;
3) Presence of features (from SLE or other chronic disease) that a-priori suggest that the subject benefits from other therapies than that suggested or allowable by the study protocol;
These disease features include but are not limited to severe, progressive, or uncontrolled hepatic, hematologic, gastrointestinal, metabolic, endocrine, pulmonary, cardiac or neurologic disease.
4) History of other kidney disease besides LN or prior to the diagnosis of SLE;
5) Need for renal replacement therapy within 2 weeks from Baseline. Subjects can have required short-term renal replacement therapy prior to Baseline, for example due to preceding acute kidney injury.
6) Infections:
a. Untreated latent or active tuberculosis (TB);
b. Chronic infections requiring treatment;
c. A subject known to be infected with Human Immunodeficiency Virus (HIV), Hepatitis B;
d. Diagnosis of any infection requiring hospitalization, parenteral antimicrobial therapy or judged to be opportunistic by the investigator within 4 weeks prior to Baseline visit;
e. Any treated infections within 2 weeks of Baseline visit;
f. History of infected joint prosthesis with prosthesis still in situ;
7) Blood dyscrasias, including:
a. Hemoglobin <8.5 g/dL or Hematocrit <22%;
b. White Blood Cell count <2.6 x 109/L;
c. Neutrophil count <1.2 x 109/L;
d. Platelet count <100 x 109/L;
e. Lymphocyte count <0.5 x 109/L.
8) Estimated glomerular filtration rate [GFR] <40 mL/min/1.73 m2 calculated using the CKiD U25 equation5,5a,5b,5c (see Appendix 4);
9) Aspartate aminotransferase (AST) or alanine aminotransferase (ALT) >1.5 times the upper limit of normal;
10) Vaccinated or exposed to a live or attenuated vaccine within the 4 weeks prior to Baseline visit;
11) History or current symptoms suggestive of lymphoproliferative disorders (e.g., Epstein Barr Virus [EBV] related lymphoproliferative disorder, lymphoma, leukemia, myeloproliferative disorders, or multiple myeloma);
12) Current malignancy or history of any malignancy with the exception of adequate treated or excised basal cell or squamous cell or cervical cancer in situ;
13) Recent (within 4 weeks prior to Baseline visit) significant trauma or major surgery;
14) Herbal supplements with pharmaceutical properties must be discontinued at least 1 week prior to Baseline visit, unless there are sufficient data available regarding the duration of an herbal medication’s pharmacokinetic and pharmacodynamic effects to allow a shorter or longer washout to be specified (e.g., 5 half-lives).
15) Oral or intravenous cyclophosphamide must be discontinued 12 weeks prior to Baseline visit
16) Use of prohibited prescription medication as listed in Appendix 3 within the specified time frame prior to Baseline visit
17) Participation in other studies involving investigational drug(s) within 4 weeks or 5 half-lives (whichever is longer) prior to Baseline visit and/or during study participation; Exposure to investigational biologics should be discussed with the Sponsor.
18) Pregnant female subjects; breastfeeding female subjects; male subjects with partners currently pregnant; male subjects able to father children and female subjects of childbearing potential who are unwilling or unable to use two highly effective methods of contraception or are abstinent (see Section 4.4.) for the duration of the study;
19) Other severe acute or chronic medical or psychiatric condition or laboratory abnormality that may increase the risk associated with study participation or investigational product administration or may interfere with the interpretation of study results and, in the judgment of the investigator, would make the subject inappropriate for entry into this study.
D. INVESTIGATIONAL AND REFERENCE THERAPY
Mycophenolate mofetil (MMF) in tablet and/or capsule form will be taken twice daily by mouth, approximately 12 hours apart. The investigational therapy will be personalized using precision dosing of MMF for subjects randomized to the MMFPK arm; or based on current standard of care (SOC), i.e., based on subject body-surface area (MMFBSA arm) this dose will be about 600 mg/m2 due to limitations imposed by the tablet or capsule strengths. MMF tablets (500 mg/ tab) and/or MMF capsules (250 mg/ cap) and/or identical placebo capsules will be used to ensure double-blinding of treatment.
The decision whether, or not, a subject is treated with MMF occurs prior to the enrollment of the patient into the study.
E. STUDY DESIGN
This is a randomized, double-blind, active comparator study of pediatric subjects (8 to < 21 years of age) with proliferative LN, with or without concurrent epimembranous immune deposits diagnosed by kidney biopsy as per ISN/RPS Classification for LN4.
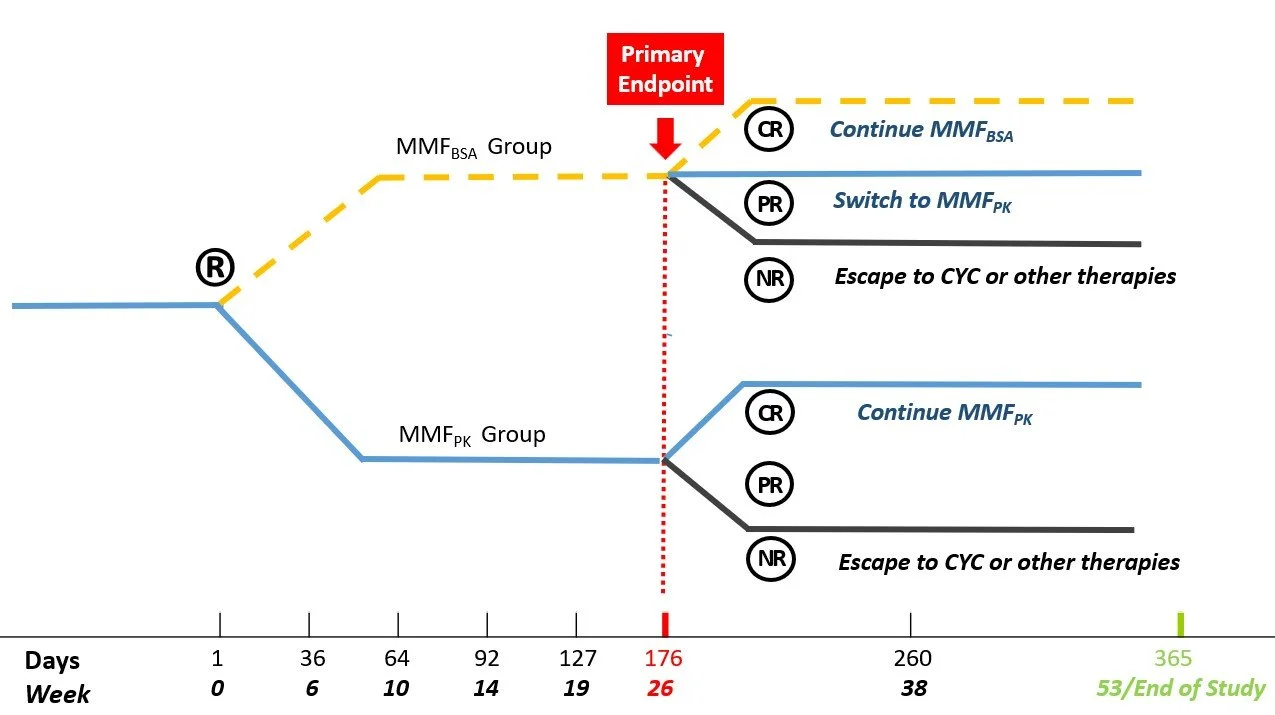
A schematic of the study design is shown in Figure 1. LN courses are defined in section 7.2.
Eligible patients enrolled in the study will be randomized (1:1) at baseline to the 53-week double-blind, active comparator 2-part study to receive either MMFPK or MMFBSA. Subjects who are partial renal responders (PRR) to MMFBSA at week 26, will cross over to the MMFPK arm. Complete renal responders (CRR) at week 26 in MMFBSA arm will continue to be treated with MMFBSA. Subjects with at least a PRR (or even CRR) in the MMFPK arm at week 26 will remain in the MMFPK arm and continue to receive MMF dosage targeting MPA-AUC0-12 > 60-70 mg*h/l. Subjects whose LN fails to respond to therapy by week 26 will be discontinued from the study interventions to receive LN treatment as per their local physician’s decision. Subjects who experience a single episode of a LN flare during Part 1 of the study or fulfill other criteria for discontinuation from the study intervention, will also receive LN treatment as per their local physician’s decision.
Figure 1: Study Design
Adherence to oral MMF will be supported by MPA measurements. Further, a smart phone app will be used for medication reminders. Study drug intake will be logged using a drug diary; and drug supply returned will be counted. MMF (study drug) will be provided by GENENTECH Inc.
Background corticosteroids use (oral, IV) will be standardized (see Section 3.2).
Subjects who are discontinued from the study intervention will continue to be monitored for a total of 53 weeks in the study. Monitoring visits occur at weeks 26 and 53.
Reasons for discontinuation from the study intervention include: failure to achieve at least PRR or occurrence of LN flare in Part 1, severe extra-renal of cSLE that necessitates use of not allowed medications (see Section 5.9), use of medications not allowed during the study for other reasons but disease flare; intolerance to study drug at the appropriate dose based on BSA (MMFBSA) or MPA- exposure (MMFPK), need for renal replacement therapy; failure to adhere to study procedures as detailed in the Schedule of Activities (SOA). A full list of criteria to be discontinued from the active study intervention is provided in (see Section 3.4.).
The Centralized Coordinating Center (CCC) will include providing feedback about the status (PRR, CRR, non-responder) and recommended dose of corticosteroids (CS).
Approximately 105 subjects will be randomized. The duration of subject participation among those who complete the study is expected to be approximately 53 weeks. The study will be conducted over a period of approximately 5 years. Approximately 20 investigative sites located in the United States and Canada are expected to participate in the study. The duration of the study may be extended, and additional investigator sites may be added depending on the observed rate of recruitment.
F. STATISTICAL ANALYSIS
For the primary endpoint, superiority of MMFPK over MMFBSA for the percentage of subjects with clinical remission of LN (PRR, CRR) at the end of Part 1 of will be tested using the analytical approach for the binomial outcome, such as Chi-square test, Fisher’s Exact, or a Bayesian’s approach with non- informative Beta prior for the binary probability parameter. The choice of the approach will be determined by the distribution of the data. Subjects who discontinued from the treatment due to any reason, will be considered as being non-responders.
All secondary and exploratory efficacy endpoints that are collected will be analyzed by treatment group. For the binary secondary or exploratory endpoints, the analytical approach for the binary outcome, as used for the primary analysis, will be performed. For the continuous exploratory endpoints, including change from baseline in PROMIS scores or RAIL scores, a mixed-effect model with repeated measures will be applied. The Kaplan-Meier plots will be generated for the exploratory endpoints of time to CRR and time to PRR. Since no measures are collected from the discontinued subject, the analyses will take an inverse propensity weighting (IPW) approach6 where the propensity for subjects to be discontinued from each arm are estimated using logistic regression modeling or the covariate balance propensity score method. The IPW approach will create pseudo subject sample that represents the original randomized subject cohort at the beginning of the study, and therefore addresses the missing data issue due to the subject discontinuation. Descriptive/summary statistics for all endpoints, with 95% CI for treatment difference, will be provided. Baseline demographic characteristics, primary and major secondary endpoints will also be summarized overall and by age (<12 years vs > 12 years), gender and race (white vs. non-white).
Safety analysis will be performed on all subjects who received at least one dose of study drug. Safety data will be subject to clinical review and summarized by appropriate descriptive statistics.
G. SCHEDULE OF ACTIVITIES
The schedule of activities table provides an overview of the protocol visits and procedures. Please refer to Section 6 (Study Procedures) and Section 7 (Assessments) of the protocol for detailed information on each procedure and assessment required for compliance with the protocol. The investigator may schedule visits (unplanned visits) in addition to those listed on the schedule of activities, in order to perform evaluations or assessments required to protect the well-being of the subject.
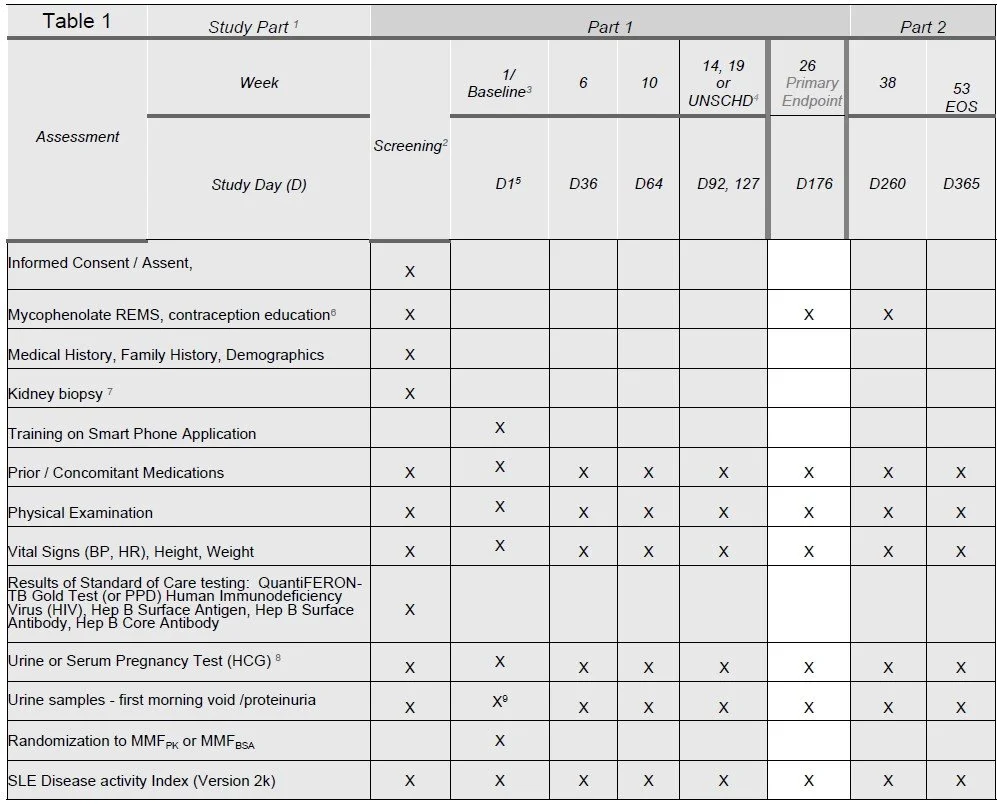
1 Visit windows are +7 days in Part 1; + 14 days in Part 2
2 Serious adverse events (SAEs) are captured from the time of informed consent at Screening; adverse events (AEs) are captured following first dose of investigational product.
3 Day 1 visit occur within 30 days of the first screening visit activity. If > 30 days has elapsed, all screening procedures and assessments must be repeated. Sponsor approval must be obtained prior to re-screening a subject
4 For unscheduled visits complete assessments listed for perceived cSLE flares or SAEs
5 If randomization does not occur at baseline for the patient, the date of randomization will be become D1.
6 Mycophenolate REMS (Risk evaluation and mitigation strategy) program and contraceptive education is for female subjects of reproductive potential only
7 Renal biopsy performed as per standard of clinical care within 90 days of enrollment
8 Urine pregnancy testing is required only for females who are of childbearing potential. Pregnancy testing will be performed locally. Pregnancy testing may be repeated more frequently if required by local practices, if a menstrual cycle is missed, or if potential pregnancy is otherwise suspected. A positive urine pregnancy test will be confirmed by a blood (serum) pregnancy test performed. A contraceptive check also will be performed to confirm that contraception, if assigned, is being used consistently and correctly. Childbearing status also will be checked and contraception implemented in subjects who mature physically and behaviorally during the conduct of the study
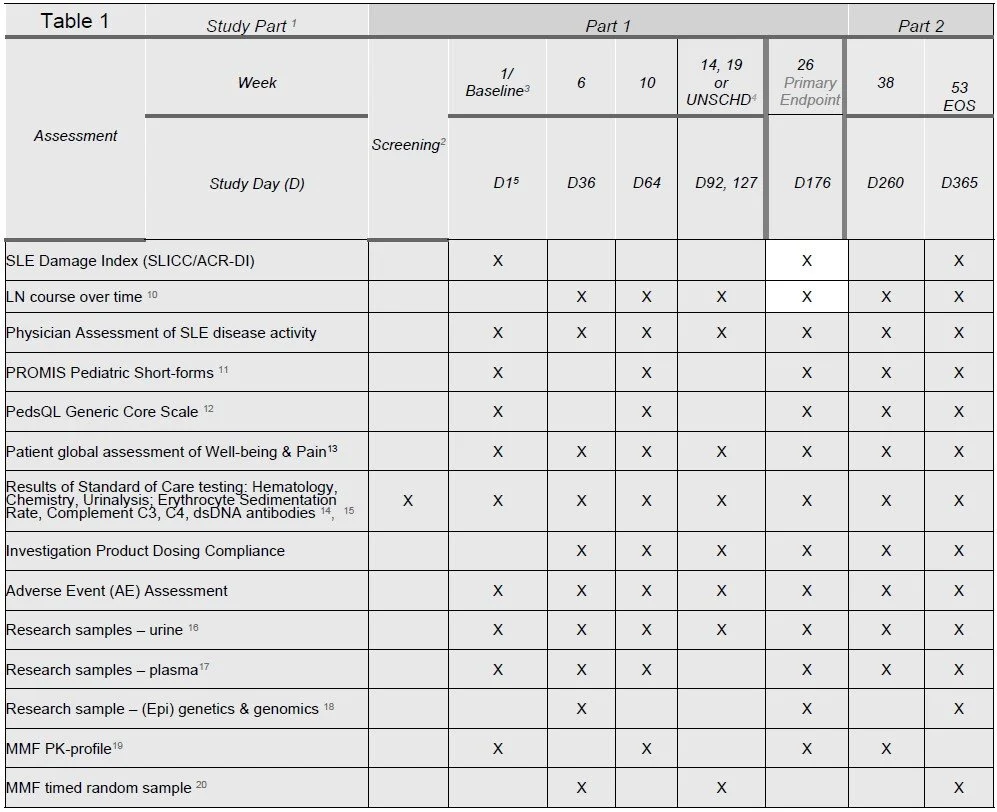
10 Partial renal response, complete renal response, non-response, lupus nephritis flare
11 At baseline (Day 1), the end of Part 1 and Part 2 the PROMIS Quality of Life short forms will be completed by the subject. If the subject is unable parent/proxy forms can be used.
12 Parent/Proxy will complete the Pediatric Quality of Life (PedsQL) Generic Core Scale
13 Parent/Proxy completes the global well-being and pain assessments for subjects under 12 years of age at study start. For subjects over 12 years of age, patient report is preferred is preferred.
14 Hematology includes: Hemoglobin, white blood cells, neutrophils (%, absolute), lymphocytes (%, absolute), monocytes, and platelets. Chemistry includes: Sodium, potassium, bicarbonate, chloride, blood urea nitrogen, creatinine, glucose, alanine aminotransferase (ALT), aspartate aminotransferase (AST), albumin. Urinalysisincludes: specific gravity, pH, protein, glucose, ketones, blood and leukocyte esterase, urine microscopy (only if dipstick positive for blood or protein, or if clinically indicated).
15 CBC, CMP, ESR, complement C3 and C4 levels and dsDNA only need to be measured at most 1 time every 2 weeks for the purpose of the study
16 10 mL of urine for biomarker testing and biobanking – from 1st morning void urine sample
17 6 mL of plasma for research
18 2.5 mL PAXgene tube and 3 mL EDTA tube
19 3-hour abbreviated PK-profile: trough, 20 min., 1 and 3 hours; additional home or on-site PK-profiling will be completed as needed
20 Additional random home testing may requested by the CCC for concerns that < 80% of medication doses are taken and after certain events 7.5.2